Stories
Lloyd Trotman: Transforming Views of Prostate Cancer
Lloyd Trotman found his calling as an experimental scientist during a “very intense” organic chemistry course as an undergraduate at the University of Zurich in Switzerland.

Lloyd Trotman
(Photo: Constance Brukin/CSHL)
“You had to do this combination of good hands-on execution of techniques and reading about what could be happening, and analyzing what you were doing … and then discussing with a mentor what it could mean,” he recalls. “People in that lab were essentially there around the clock, so I immediately got hooked on that lifestyle—of really living through experimentation.”
He later decided to apply his passion to tackling questions of disease. Trotman has focused his career on decoding the molecular mechanisms of prostate cancer, the second most common cause of cancer death in American men (after lung cancer). His research has uncovered critical genetic regulators of the disease, and his laboratory has created an innovative system for experimentally modeling metastatic prostate cancer. By addressing a longstanding challenge in the field, this development promises to bring new understanding of the disease and better approaches to testing potential therapies.
Trotman began his Ph.D. in molecular biology in the mid-1990s. He chose to pursue research on viruses, he says, in part because “HIV research was really strong, and was starting to show some progress, but it was still a lethal disease.” Working with Urs Greber, Trotman explored how adenovirus delivers its DNA into the nucleus of a host cell—an essential step for viral replication. “It was a very rewarding experience,” he says, “to translate from observing the virus as a ‘master thief’ infecting a cell, to learning [the cellular processes] that it’s trying to exploit, and therefore, the processes that are very basic and fundamental to the cell.”
Studying in the University of Zurich’s Institute of Zoology, he was impressed by other labs’ research in fruit fly genetics, which showed him the power of whole-organism models to shed light on human diseases. Trotman moved to New York for a postdoctoral fellowship at the Sloan Kettering Institute for Cancer Research with Pier Paolo Pandolfi (now at Harvard’s Beth Israel Deaconess Medical Center). Pandolfi’s lab was pioneering the use of mouse models to study cancer, and Trotman generated new models to investigate how the inactivation of a key tumor suppressor gene, PTEN, affected the progression of prostate cancer. He also helped to elucidate the activities of the PTEN protein within the cell, as well as related pathways that contribute to cancer through uncontrolled cell survival and growth.
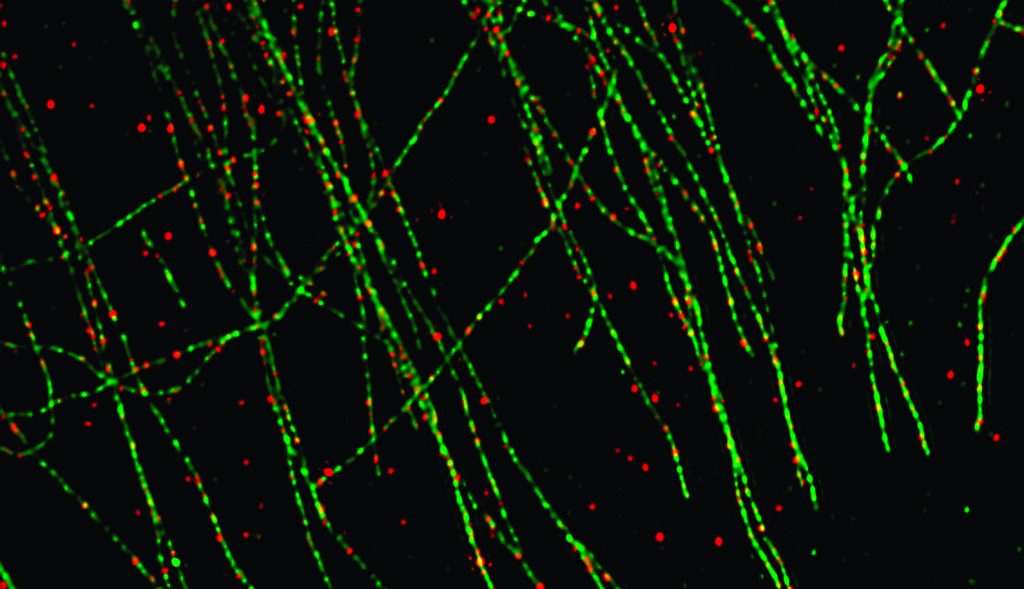
Rita Allen Foundation Scholar Lloyd Trotman and his team have used super-resolution microscopy to track the subcellular activity of the PTEN protein, a tumor suppressor that is frequently inactivated in prostate cancer. Here, red punctae mark the locations of PTEN proteins, which are associated with cellular “roads” made up of microtubules, marked in green.
(Image: Trotman Lab, CSHL)
In 2007 Trotman joined the faculty at Cold Spring Harbor Laboratory and became a Rita Allen Foundation Scholar. This was his first grant application as an independent investigator, and he still deeply appreciates that the scientists on the selection committee “were interested in what should be discovered in the future and the questions that should be asked in the future.” The award enabled his lab to devise a variety of approaches for identifying genes that interact with PTEN to influence prostate cancer outcomes—finding new factors, such as the tumor suppressor gene PHLPP1, that help determine whether the disease metastasizes and becomes lethal.
Perhaps Trotman’s most significant contribution to date has been the development of the RapidCaP system (CaP is an abbreviation for “cancer of the prostate”) for modeling metastatic prostate cancer in mice. The technique uses a modified virus to deliver cancer-promoting genes directly into the prostate, along with genes for the production of bioluminescent proteins to enable visual tracking of the disease. Compared to previous models, mice engineered using RapidCaP more faithfully reproduce the dynamics of human cancer. This makes the mice a vital tool for moving beyond hormone deprivation therapy, the long-established standard of care for prostate cancer, which Trotman says often amounts to simply “buying time” for the patient until his cancer comes back in a more lethal form.
“What has been really satisfying is that our model was the first one to generate massive amounts of metastatic disease so that we could really look at it. And when we looked at it we started to see things that were entirely unexpected.”
In addition to employing the RapidCaP system to analyze the progression and treatment of prostate cancer, Trotman and his research group are continuing to uncover the fundamental molecular mechanisms of PTEN and other cancer-associated signaling proteins. Here, Trotman explains the impetus for generating RapidCaP mouse models, and discusses how these models are reshaping scientists’ understanding of prostate cancer and approaches to therapy.
What led you to develop the RapidCaP system?
We developed it out of necessity. We always believed that genetically engineered mice were the best way to simulate human disease, but there were serious bottlenecks with the existing models. One bottleneck was that we had made mice that carried genetic mutations that very clearly made the most aggressive possible prostate cancer, but these mice did not develop metastases and they did not die of metastases.
This was a clear discrepancy between the models and the human disease, where the disease in the prostate is not a problem at all. It can be removed, but the person will still die because there is in some cases invisible, metastatic disease that will finally grow to a size that causes failures of multiple organs. We thought we were not modifying the right genes, or perhaps that the mouse was just inherently different, but then whole genome sequencing of cancer, and especially of metastatic cancer, was for the first time feasible. At that point we realized that something like half of lethal disease in patients actually had the genotype that we were modeling, so we couldn’t blame it on the genes—there was something wrong with the way we were modeling the disease.
Another thing was that the way the disease was modeled was extremely impractical; it had many drawbacks because of the technicalities of [mouse] breeding. This classic genetics-based approach generated cancer in every cell of the prostate—in the mouse that’s roughly 1 million cells—and every cell would acquire the genetic changes that should then trigger the disease. But what we think is happening in humans is that a single cell acquires mutations and then successfully multiplies and eventually causes lethal disease. So, we thought one approach to achieve this [in mice] would be to use viruses to deliver genes into the prostate so that they would infect very few cells, and these cells would be the origin of disease.
What have you learned from these new models of the disease?
It’s a bit paradoxical to think that having fewer [cancerous] cells to start with would be more likely to generate metastatic disease, as opposed to starting with 1 million cells, but this is exactly what we happened to discover. That was underappreciated … when 1 million cells become cancerous in the prostate, the environment reacts and tries to isolate the cells, but if only a few cells turn into cancer, these defenses are very weak, and these cells can escape.
So, we realized that we had a completely new way of recreating human disease, which gave us a lot more flexibility. Instead of breeding mice with different genotypes, we were introducing viruses that had specific genes, and this system is much easier and faster to re-engineer with molecular biology. On top of that, monitoring [the spread] of disease is extremely important and is very hard to achieve in humans. We could now very easily introduce marker genes that cause the cancer cells to shine through the skin, allowing us to monitor the mice as the disease evolves, and at all times get an objective analysis of what the disease is doing. In humans, you need very specific modalities that perhaps are specific for bone disease, specific for a disease that is metabolically active, or a disease that has a certain size. This is different. This marks every cell that’s cancerous.
What has been really satisfying is that our model was the first one to generate massive amounts of metastatic disease so that we could really look at it. And when we looked at it we started to see things that were entirely unexpected. The pathway that we use to drive the disease—that we know is driving the early disease stages—was completely absent in the late disease stage. In the last few years we’ve had the reward of seeing that human analysis is getting better and better, and we’ve realized that the lethal disease looks very much like what we’re describing—a disease that’s no longer driven by the pathway that originally triggered it, and no longer has key markers that are associated with the prostate. We’re reinvigorated in trying to find therapies that could kill that kind of disease because we think that’s ultimately the disease that will kill a patient.