Stories
Titia de Lange: The Complex Puzzle of Chromosome Ends
When asked to describe her research, Titia de Lange says she has been “working on the same simple question for 25 years.” That’s a modest account of a dogged dedication that has revealed information critical to understanding both genome maintenance and cancer development.
De Lange studies how cells protect the ends of their chromosomes, or telomeres, from getting chewed up by DNA repair machinery. She has examined this erosion in early-stage cancer, explaining how the loss of telomere function can generate genome instability and drive cancer progression.
While de Lange’s efforts have yielded striking advances, she would never have expected these achievements when she began her “meandering path” toward becoming a research scientist.

TITIA DE LANGE
(PHOTO: JOHN ABBOTT/THE ROCKEFELLER UNIVERSITY)
After finishing high school in the Netherlands, de Lange had to choose a career track before starting her university studies. “I decided I wanted to study chemistry, because that was the only thing I enjoyed,” she says. “But there were no women in chemistry—not just professors, but no students either. So I decided to go to biology because they offered a biochemistry track, and I thought that would be close enough.”
The “descriptive” nature of her biology training and the endless memorization of anatomical details left de Lange uninspired, until she met molecular biologist Richard Flavell (now at the Yale School of Medicine) at the University of Amsterdam. In Flavell’s laboratory, then at the National Institute for Medical Research in Mill Hill near London, de Lange completed the equivalent of a master’s thesis, helping to identify a genetic translocation implicated in a rare form of thalassemia, a disorder marked by abnormal production of hemoglobin. “That was where I first saw how science is really done,” de Lange recalls. “It was a very vibrant, competitive, international lab. It was a lot of fun, so that made me stay in science.”
She earned a Ph.D. at the Netherlands Cancer Institute, where she worked with Piet Borst to study the genetic underpinnings of variant surface glycoproteins in the parasite Trypanosoma brucei, which causes African sleeping sickness. These investigations piqued de Lange’s interest in telomeres, as the glycoprotein genes’ positions near the ends of chromosomes appeared to facilitate the duplications and transpositions that help the parasite dodge the immune system.
“The more I thought about it, the more I saw that there was a real problem with chromosome ends, and it was beginning to look like an impossible issue for nature to deal with. The problem is that we have linear chromosomes, they have ends, and those ends look like broken DNA, but the cell should not treat them like broken DNA.”
This line of research led de Lange to a postdoctoral fellowship at the University of California, San Francisco, where she examined the structure of human telomeres in the laboratory of Harold Varmus. (Varmus shared the 1989 Nobel Prize in Physiology or Medicine with J. Michael Bishop for pioneering work on cancer-causing oncogenes, and later directed the National Institutes of Health and the National Cancer Institute.) De Lange discovered that sperm cells have longer telomeres than somatic cells, and that tumor cells have notably short, unstable telomeres—findings that continue to guide telomere research.
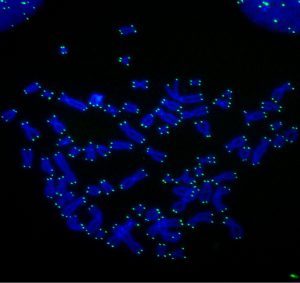
Rita Allen Foundation Scholar Titia de Lange studies how cells protect their chromosome ends, or telomeres, from damage by DNA repair mechanisms. This microscopic image shows human chromosomes labeled with a telomere-specific probe (green).
(Image: Laboratory of Cell Biology and Genetics at The Rockefeller University/PLoS Genetics)
After establishing her own laboratory at The Rockefeller University in 1990, de Lange began to focus on identifying telomere-associated proteins and assessing their roles in shielding telomeres from DNA repair processes. She was a Rita Allen Foundation Scholar from 1995 to 1998, a fruitful period for her research group. De Lange and postdoctoral fellow Bas van Steensel (now a research group leader at the Netherlands Cancer Institute) demonstrated that the human telomeric-repeat binding factor protein TRF1 regulates the length of telomeres. They also discovered that a related protein, TRF2, prevents telomeres from fusing with one another. (Agata Smogorzewska, who conducted her Ph.D. research with de Lange and became a Rita Allen Foundation Scholar in 2010, also coauthored this study and was the lead author on later work integrating the roles of TRF1 and TRF2.)
Around the same time, de Lange and her team identified an enzyme called tankyrase that interacts with TRF1 and has since been shown to release TRF1 from telomeres, allowing access by the telomere-lengthening enzyme telomerase. And a collaboration with Jack Griffith, an electron microscopy specialist at the University of North Carolina at Chapel Hill, revealed that TRF2 functions to remodel telomeres into a “loop” structure, tucking in overhanging single strands of DNA and protecting the chromosome end.
By 2005, de Lange’s work had led to the realization that a dynamic complex of six proteins, which she dubbed shelterin, function together to regulate the length of telomeres and “shelter” them from DNA damage response and repair pathways. “For the last 15 years, we’ve been doing experiments to figure out how these proteins do this, and we’re not done yet,” she says. Here, de Lange recalls the origins and progress of her investigations into telomere protection, and details emerging evidence on links between telomeres and tumors.
How did you come to focus on telomere biology?
I can’t say I had a brilliant eureka insight, but the more I thought about it, the more I saw that there was a real problem with chromosome ends, and it was beginning to look like an impossible issue for nature to deal with. The problem is that we have linear chromosomes, they have ends, and those ends look like broken DNA, but the cell should not treat them like broken DNA.
As we were learning about how cells respond to broken DNA, the problem of how telomeres could prevent all this became more complex and mysterious. My model was that there would be proteins [associated] with telomeres that would fulfill this function. How, I didn’t know, but I thought, “There have to be some proteins there that do this.”
What has your research revealed about how cells maintain their telomeres?
At first, this project was so risky that nobody in my lab wanted to work on it, so the students and postdocs worked on other things, and I worked with a technician on finding a protein [TRF1] that bound to telomeric DNA. It took five years, and it was a terrible project—once we identified it, purifying this protein took the whole supply budget of my single NIH grant. But once we got it, in the next five to seven years we identified a number of other proteins. Other people started working on this as well, and by 2005 I proposed that all these proteins formed a single complex that I called shelterin, for the sheltering of chromosome ends.
Now we know which protein has which task. It’s a beautiful little complex. It only has six members, but it is able to repress six very different pathways that cells can activate at DNA ends. One issue is that when the telomere is linear, it has DNA ends that cells can respond to. So the complex folds back the telomere; it tucks the DNA end away so that cells can’t see it. This is an elegant architectural solution. I would say that the identification of the shelterin complex and the understanding of how it solves this end protection problem—that’s partly due to my contribution.
What questions do you want to answer going forward?
With regard to the end protection problem, we are not there yet. We understand how two of the six pathways are repressed, and we’re making progress toward the others, but there’s a ways to go to understand mechanistically how this really works.
In addition, I think the field will more and more turn its attention to what happens to telomeres during tumorigenesis. We know that telomeres shorten in normal, even somatic [non-gamete] cells, because telomerase is switched off. That shortening provides our tissues with a replication barrier. A cell can only divide until it runs out of telomeric DNA, and then it has to stop—that’s a good way to prevent cancer from arising. It works very well, but cancer always finds ways around tumor-suppressor pathways. When cells continue to divide even though their telomeres are too short, you get genome instability, which can actually promote tumorigenesis—this is referred to as telomere crisis.
We now realize that telomere crisis is very frequent in human cancers at an early stage, and that some of the massive genome rearrangements that we observe in full-blown tumors can be ascribed to telomere dysfunction. So my lab is increasingly focused on understanding the actual consequences of telomere dysfunction in the early stages of tumorigenesis. I would like to better understand the exact genomic signatures of telomere crisis so that we can identify which tumors have lived through that misery in their proliferative past, and perhaps this can help us with prognosis, diagnosis and so on.